Gigantyzm to nietypowo duży wzrost. Choroba ma dwie odmiany – jedna z nich występuje u dzieci, druga – u dorosłych. Gigantyzm powodowany jest przez nadczynność przedniego płata przysadki i wzmożone wydzielanie hormonu wzrostu, co doprowadza do tak zwanego wzrostu olbrzymiego. Choroba rozwija się w wieku młodocianym, kiedy nie doszło jeszcze do zrastania się nasad kości i skostnienia chrząstek wzrostowych. Jeśli wydzielanie hormonu wzrostu ustanie po osiągnięciu okresu pokwitania, to wzrost olbrzymi nie będzie łączył się z cechami akromegalii. Jeśli nadal będzie wytwarzany – do gigantyzmu dołączy akromegalia - o której piszę niżej.
1. Przyczyny i objawy gigantyzmu
Najczęstszą przyczyną wydzielania zbyt dużej ilości hormonu wzrostu jest niezłośliwy guz przysadki. Inne choroby wpływające na wystąpienie wzrostu olbrzymiego to:
zespół Carney'a,
syndrom McCune-Albrighta,
mnoga gruczolakowatość typu 1,
nerwiakowłókniakowatość.
Gruczolak lub przerost komórek kwasochłonnych przedniego płata przysadki powodują wydzielanie w nadmiarze hormonu wzrostu. U dzieci powoduje to zwiększenie wzrostu, wpływa także na narządy wewnętrzne. U dorosłych – wzrost się nie zmienia, tylko powiększają i pogrubiają się kości, chrząstki i tkanka łączna, również narządy mogą zmienić swój rozmiar. Dorośli chorują najczęściej pomiędzy 30. a 45. rokiem życia. Inne objawy wzrostu olbrzymiego to:
opóźnione pokwitanie,
podwójne widzenie lub trudności z widzeniem bocznym,
wyraźnie zarysowana, wystająca szczęka,
bóle głowy,
nadmierne pocenie się,
nieregularne miesiączki,
duże dłonie i stopy z grubymi palcami,
wydzielanie mleka z piersi,
zgrubienie rysów,
osłabienie.
Wystąpienie powyższych objawów powinno skłonić do wizyty u lekarza, by ustalić przyczynę nietypowo dużego wzrostu. W celu rozpoznania choroby przeprowadza się następujące badania:
tomografię komputerową lub obrazowanie metodą rezonansu magnetycznego – pomagają wykryć ewentualnego guza przysadki,
badanie poziomu hormonu wzrostu,
badanie poziomu prolaktyny – wysoki wskazuje na gigantyzm,
badanie insulinopodobnego czynnika wzrostu – jego wysoki poziom może być oznaką wzrostu olbrzymiego.
Uszkodzenie przysadki może spowodować niski poziom kortyzolu, estradiolu (u dziewcząt), testosteronu (u chłopców), a także hormonu tarczycy.
2. Leczenie wzrostu olbrzymiego
W przypadku wystąpienia guza przysadki z wyraźnie zaznaczonymi brzegami, operacja jego wycięcia to dobra i często stosowana opcja leczenia. U wielu chorych zabieg chirurgiczny przynosi pożądane rezultaty. Niekiedy jednak chirurg nie jest w stanie całkowicie usunąć guza, dlatego korzysta się także z innych metod leczenia. Najskuteczniejsza jest terapia somatostatyną, która zmniejsza wydzielanie hormonu wzrostu. Stosowani są także agoniści dopaminy, ale takie leczenie jest mniej efektywne. W celu przywrócenia normalnego poziomu hormonu wzrostu, lekarze sięgają niekiedy po radioterapię. Jednak uzyskanie optymalnych efektów leczenia trwa 5-10 lat. Wielu ekspertów korzysta z radioterapii wtedy, gdy operacja i przyjmowanie leków nie zdają egzaminu.
Bibliografia
Herrmann F., Lohmann T., Muller P., Endokrynologia w praktyce klinicznej. Diagnostyka i leczenie, Wydawnictwo Lekarskie PZWL, Warszawa 2009, ISBN 978-83-200-3835-4
Syrenicz A. Endokrynologia w codziennej praktyce lekarskiej, Pomorska Akademia Medyczna, Szczecin 2009, ISBN 978-83-61517-14-6
Burch W.M. Endokrynologia, Urban & Partner, Wrocław 1996, ISBN 83-85842-51-9
Szczeklik A. (red.), Choroby wewnętrzne, Medycyna Praktyczna, Kraków 2011, ISBN 978-83-7430-289-0
Akromegalia
Co to jest i jakie są przyczyny?
Akromegalia jest chorobą przewlekłą związaną z nadmiernym wydzielaniem hormonu wzrostu. Charakteryzuje się postępującą stopniowo zmianą wyglądu zewnętrznego oraz wieloma powikłaniami narządowymi. Akromegalia występuje u osób dorosłych, u których zakończony został proces wzrastania kości. Natomiast u dzieci i młodzieży nadmiar hormonu wzrostu prowadzi do tzw. gigantyzmu (nadmiernego wzrostu ciała).
Hormon wzrostu jest wytwarzany przez wyspecjalizowane komórki przysadki, gruczołu położonego w środkowym dole czaszkowym. U dzieci hormon wzrostu jest konieczny dla prawidłowego wzrastania, u dorosłych wpływa na metabolizm w organizmie, czynność kości i mięśni.
Hormon wzrostu prowadzi do wytwarzania przez wątrobę czynników wzrostowych, głównie IGF-1 (insulinopodobny czynnik wzrostu; insulin-like growth factor-1, inaczej nazywany somatomedyną C). Czynniki wzrostowe są odpowiedzialne za efekty działania hormonu wzrostu.
Jak często występuje akromegalia?
Akromegalia występuje rzadko. Corocznie chorobę stwierdza się u około 3–4 nowych osób na milion. Ocenia się, że w Polsce na akromegalię choruje ok. 2000 osób. Chorobę obserwuje się dwukrotnie częściej u kobiet.
Najczęstszą przyczyną akromegalii jest gruczolak (łagodny guz) przysadki.
Jak się objawia akromegalia?
Zmiany wyglądu zewnętrznego typowe dla akromegalii rozwijają się powoli i są przez długi okres niedostrzegalnie dla chorego. Zwykle od pojawienia się pierwszych objawów choroby do rozpoznania akromegalii upływa kilka lat. Dochodzi do powiększenia rąk i stóp, co zmusza chorych w okresie kilku lat do zmiany rozmiaru noszonych butów i rękawiczek na większy (u kobiet pierwszym sygnałem może być konieczność zmiany rozmiaru pierścionków). Powiększeniu ulega również twarzoczaszka - typowy dla akromegalii jest szeroki i gruby nos, powiększenie żuchwy z szerokimi szparami między zębami, wydatne łuki brwiowe. Chorzy często skarżą się na nadmierną potliwość.
Nadmierne wydzielanie hormonu wzrostu prowadzi również do rozwoju powikłań w wielu układach:
krążenia (nadciśnienie tętnicze, niewydolność serca),
oddechowym (uporczywe chrapanie i okresy bezdechu w czasie snu),
pokarmowym (zaparcia i polipy jelita grubego),
wewnątrzwydzielniczym (cukrzyca, powiększenie tarczycy i nadczynność tarczycy),
kostno-stawowym (choroba zwyrodnieniowa stawów),
nerwowym (bóle głowy, zespół cieśni nadgarstka).
Miejscowe powiększanie się guza może powodować ucisk na zdrową część przysadki i objawy jej niedoczynności (niedoboru hormonów przysadki). Ponadto duże guzy (makrogruczolaki) mogą powodować bóle głowy i zaburzenia widzenia (zawężenie pola widzenia).
Co robić w razie wystąpienia objawów?
W razie pojawienia się objawów choroby należy zgłosić się do lekarza endokrynologa (specjalisty w dziedzinie gruczołów wydzielania wewnętrznego i hormonów), który zleci wykonanie badań potwierdzających rozpoznanie.
Jak lekarz ustala diagnozę?
Przy podejrzeniu akromegalii, lekarz w pierwszej kolejności wykonuje badania krwi obejmujące: ocenę stężenia IGF-1 (u chorych poziom tego czynnika wzrostowego jest podwyższony); test hamowania wydzielania hormonu wzrostu po podaniu glukozy (u osób zdrowych wysokie stężenia cukru we krwi hamuje wydzielanie hormonu wzrostu, u chorych z akromegalią obserwuje się brak hamowania).
Po potwierdzeniu rozpoznania lekarz zleca badania obrazowe przysadki (rezonans magnetyczny lub tomografię komputerową) w celu uwidocznienia guza i oceny jego wielkości. Zazwyczaj u chorych z akromegalią stwierdza się makrogruczolaki (guzy >1 cm średnicy). W każdym przypadku makrogruczolaka niezbędna jest konsultacja okulisty (specjalisty chorób oczu). Ponadto w przypadku makrogruczolaka lekarz wykonuje badania krwi w celu wykluczenia niedoczynności przysadki (patrz: Niedoczynność przysadki).
Jakie są sposoby leczenia?
Celem leczenia guzów wydzielających hormon wzrostu jest usunięcie (ewentualnie zmniejszenie) guza oraz obniżenie stężenia hormonu wzrostu i IGF-1 do wartości prawidłowych.
Najlepszą metodą leczenia jest operacja usunięcia guza przez nos i zatokę klinową, wykonywana w wyspecjalizowanych ośrodkach neurochirurgicznych. Dzięki opracowaniu takiej drogi dojścia do przysadki, zabieg operacyjny wykonuje się bez otwierania czaszki, co zmniejsza ryzyko możliwych powikłań. Takie leczenie może prowadzić do całkowitego wyleczenia (chory nie wymaga dodatkowego leczenia guza przysadki). Niestety całkowite usunięcie dużego guza może być trudne i niekiedy po zabiegu konieczne jest leczenie dodatkowe lub ponowne operacje.
Istnieją leki obniżające stężenie hormonu wzrostu i zmniejszające wielkość guza (ale nie prowadzące do jego zniknięcia). Najczęściej stosuje się wstrzyknięcia analogów somatostatyny. Analogi somatostatyny można stosować przed planowaną operacją lub po niecałkowitym usunięciu gruczolaka. W razie ich nieskuteczności czasami rozważa się dołączenie leków dopaminergicznych lub terapię antagonistą receptora hormonu wzrostu. W wyjątkowych sytuacjach (leczenie operacyjne nieskuteczne lub niemożliwe do przeprowadzenia , nieskuteczne leczenie farmakologiczne) wykonuje się radioterapię. Obecne leczenie radioterapią polega na zastosowaniu pojedynczej dawki promieniowania lub serii małych dawek promieniowania nakierowanych wybiórczo na gruczolaka. Leczenie takie pozwala na zmniejszenie rozmiarów guza i uzyskanie prawidłowych stężeń hormonu wzrostu, ale ostateczny efekt leczenia uzyskuje się dopiero po kilku, lub nawet kilkunastu latach. Zaletą radioterapii jest trwałość uzyskanego efektu leczenia, zaś minusem - poważne działania niepożądane, jak niedoczynność przysadki (nabyty brak wytwarzania jednego lub wielu hormonów przysadki) czy popromienne uszkodzenie nerwu wzrokowego i ślepota (zwykle jednego oka).
Czy możliwe jest całkowite wyleczenie?
Należy podkreślić, że wczesne rozpoznanie akromegalii pozwala na skuteczne leczenie objawów i zapobiega rozwojowi powikłań.
Leczenie operacyjne jest jedyną metodą, która może prowadzić do całkowitego wyleczenia. Leczenie farmakologiczne jest przewlekłe i pozwala jedynie na spowolnienie i kontrolowanie przebiegu choroby.
Co trzeba robić po zakończeniu leczenia?
Chory wymaga stałej opieki w poradni endokrynologicznej. Po zakończeniu leczenia wskazana jest okresowa kontrola badań krwi i wykonywanie kontrolnych badań obrazowych.
Co robić, aby uniknąć zachorowania?
Sposoby zapobiegania wystąpieniu akromegalii nie są obecnie znane.
http://www.mp.pl/pac...595,akromegalia
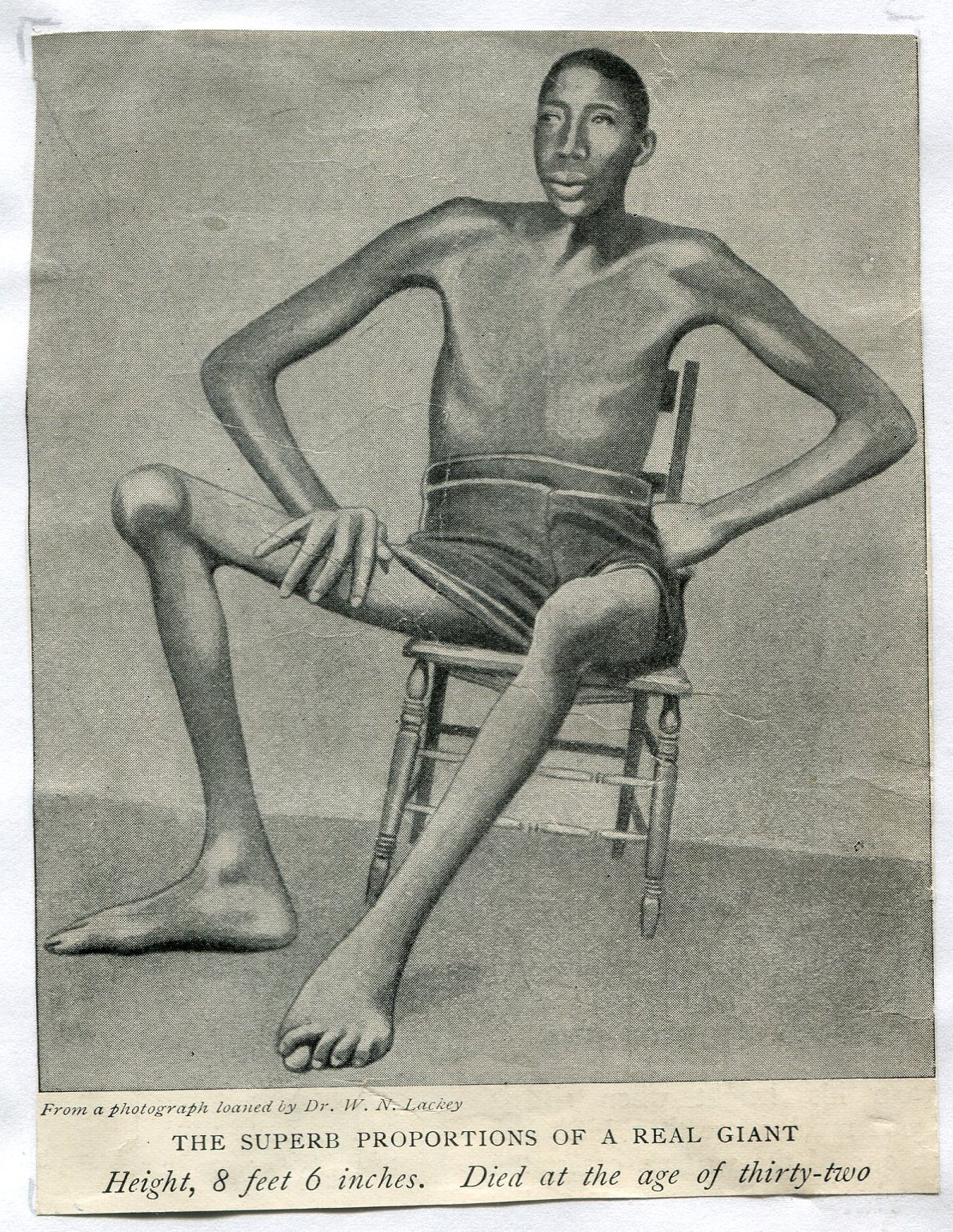
Kilka zdjęć ludzi, którzy chorowali na gigantyzm:
Matthew McGrory (ur. 17 maja 1973 w West Chester, zm. 9 sierpnia 2005 w Los Angeles) - amerykański aktor znany głównie ze swojego wysokiego wzrostu - 2,29 m.
McGrory urodził się w West Chester w Pensylwanii i uczył się w szkole prawniczej Widener University, lecz zainteresował się aktorstwem grywając olbrzymów (np. w filmie Tima Burtona Duża ryba, ang. Big Fish). Mieszkał z Melissą Davis w Sherman Oaks w Kalifornii. Zmarł z przyczyn naturalnych w Los Angeles w wieku 32 lat.


Robert Pershing Wadlow (ur. 22 lutego 1918 w Alton, Illinois, USA, zm. 15 lipca 1940, Manistee, Michigan, USA) – najwyższy znany człowiek w historii.
Jego rodzicami byli Addie i Harold Wadlowowie. Podczas narodzin ważył niespełna cztery kilogramy. W wieku sześciu miesięcy osiągnął wagę trzynastu i pół kilograma, zaś po roku - trzydziestu kilogramów. W 5 roku życia mierzył 155 cm wzrostu, w trzeciej klasie szkoły podstawowej przerósł nauczyciela. Gdy miał 13 lat, mierzył już 224 cm. Już w wieku 18 lat urósł do 254 cm. W wieku 22 lat, przed śmiercią, osiągnął wzrost 272 cm[1] i wagę 222 kg.
4 lipca 1940 podczas występu doznał obtarcia kostki, co doprowadziło do powstania pęcherza, który później uległ zakażeniu. Mimo starań lekarzy Robert Wadlow zmarł 15 lipca 1940 o godzinie 1:30 w nocy w Manistee w stanie Michigan[2]. Pochowano go na cmentarzu w rodzinnym Alton nad rzeką Missisipi.